

Abstract
Introduction: Primary immunodeficiency diseases (PID) are caused by gene defects that impair function of the innate or adaptive immune systems. An increasing number of patients have been identified with a causative monogenic defect. More than 300 different genetic defects have been found [J Clin Immunol (2018) 38:96-128]. The NFKB1 gene is strongly associated with PID. Heterozygous variants of NFKB1 cause a progressive defect in formation of immunoglobulin-producing B cells [Cell (2017) 168:37-57]. Here, we introduce a new NFKB1 mutation.
Methods: A patient with a 20-year history of diarrhea was recently hospitalized due to three months of interrupted fever. Chest computed tomography (CT) showed bilateral pneumonia, splenomegaly, and retro-peritoneal lymphadenopathy. We highly suspect that he has primary immunodeficiency and collected blood samples from all family members to identify the gene mutation.
Family history. The patient's father (I.2) died early and his mother (I.1) died of cerebral infarction a few months ago. The patient has two brothers and two sisters. One brother (II.1) died of tuberculosis, the other (II.4) is healthy. One sister (II.3) died of stomach cancer, the other (II.2) has a history of left breast cancer and has received chemotherapy three times. His son (III.1) is clinically asymptomatic. His wife (II.6) is healthy (Figure 1).
Blood samples. We evaluated complement components and quantified immunoglobulin levels of the family members, and determined the B cell ratio of the patient. Next, we performed whole exon sequencing by next-generation sequencing. We also predicted the protein structure of the mutant gene.
Results: The patient has severely decreased levels of serum IgG, IgA, and IgM. Unexpectedly, his son has moderately reduced IgG levels, while others' immunoglobulin is normal. The patient's lymphocyte subgroups revealed a high ratio of CD3+, CD3+/CD8+ lymphocytes and low ratio of CD19+, CD56+CD16+ lymphocytes, which suggests a decreased proportion of B lymphocytes (Table 1).
Next-generation sequencing revealed all known gene mutations of this family. Using Phenolyzer software (a tool that uses prior information to implicate genes involved in diseases), we selected three candidate genes: RAG1, C2, and NFKB1. The patient (II.5), his sister (II.2), and his son (III.1) all have a heterozygous mutation of RAG1. Thus, we ruled out RAG1, as it does not conform to Mendel's laws in this family. C2 was also excluded due to the low haploinsufficiency score (0.178). Interestingly, the patient (II.5) and his son (III.1) both have a heterozygous mutation of NFKB1, while others do not. NFKB1 shows a high haploinsufficiency score (0.945), suggesting that the single functional copy of the gene may not produce enough protein. Thus, we hypothesize that NFKB1 is the disease-causing gene in this family.
Further investigation revealed a heterozygous NFKB1 frame shift mutation (c.2053delG: p.G685fs) in the patient and his son. Other family members possess wild-type NFKB1. The novel frameshift mutation influences three transcriptomes with a similar coding sequence to the NFKB1 gene. Sanger sequencing verified the results.
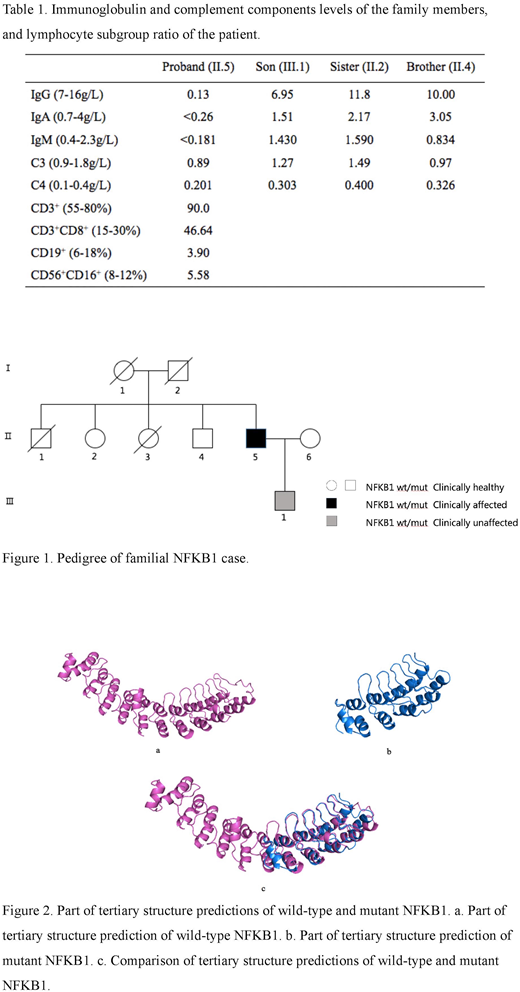
The NFKB1 gene consists of four regions: Rel homology, glycine-rich, ankyrin repeats, and DEATH domain. Our prediction of the protein structure suggests that the frameshift mutation occurred in the ankyrin repeats region. Studies have shown that large deletions in the ankyrin repeats region may cause deficiency in class-switched memory B cell generation. This mutation results in a loss of 283 amino acids and addition of 40 new amino acids.
Prediction of the tertiary structure illustrated that the coding protein is terminated early. The mutation results in loss of some helixes and formation of a new helix at the C-terminal. This is a novel mutation of NFKB1 that has not previously been reported in PID, and which forms a new protein structure (Figure 2).
Conclusions: Our findings broaden the scope of phenotypes caused by mutations in NFKB1. We suspect that this heterozygous mutation of NFKB1 may lead to fewer immunoglobulins produced.Onset was delayed for this patient, at the age of 20. His son is 25 years old now, with moderately reduced levels of IgG but without symptoms. We suspect that he may be ill in the future and recommended that he seek genetic counseling when he is ready to have a child.
No relevant conflicts of interest to declare.

This feature is available to Subscribers Only
Sign In or Create an Account Close Modal